Software
pyCRAC software
pyCRAC is a comprehensive software package designed for the analysis of high-throughput sequencing data derived from CRAC (Cross-linking and Analysis of cDNA) and CLIP (Cross-linking and Immunoprecipitation) experiments. It provides a suite of tools for read preprocessing, mapping, and downstream analysis to identify RNA-protein interaction sites with high resolution.
Note that the latest version of pyCRAC is compatible with Python 3. Version 2.0 is on the way, which will feature massive improvements in performance and usability.
Code repository: https://git.ecdf.ed.ac.uk/sgrannem/pycrac
Installation
Install the latest stable release from PyPI:
pip install pyCRACInstall from Bioconda:
conda install bioconda::pycracInstall the latest development version from GitLab:
git clone https://git.ecdf.ed.ac.uk/sgrannem/pycrac.git
cd pycrac
pip install .A software package without bugs does not exist. Please check the repository regularly for bug fixes and other updates. Feedback is also much appreciated!!
Please cite the following paper when using the package for your data analyses:
Shaun Webb, Ralph D. Hector, Grzegorz Kudla and Sander Granneman.
PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast.
Genome Biology 2014;15:R8; DOI: 10.1186/gb-2014-15-1-r8
Here are a few examples of how to use GTF2 parser in your python programs.
from pyCRAC.Parsers import GTF2
from pyCRAC.Methods import translate
import numpy as np
import re
gtf = GTF2.Parse_GTF()
gtf.read_GTF("Saccharomyces_cerevisiae.R64-1-1.75_1.2.gtf",transcripts=False)
gtf.read_FASTA("Saccharomyces_cerevisiae.R64-1-1.75.fa")
# Get genomic coordinates for gene RPL7A
gene = "RPL7A"
coordinates = gtf.chromosomeGeneCoordIterator("I")
See the full documentation in the package.
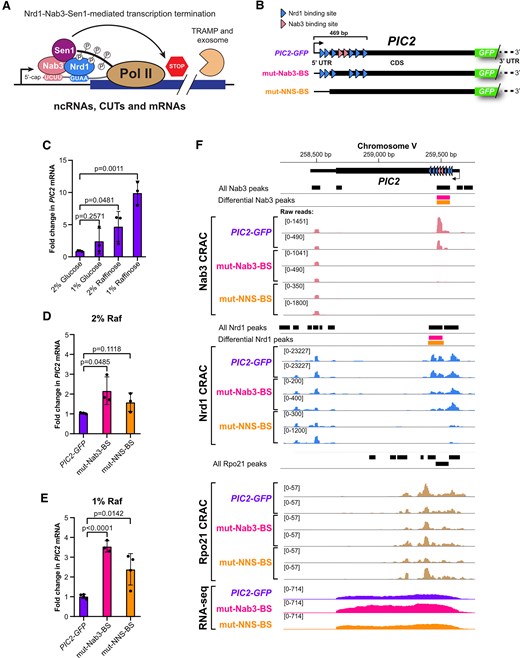
DBPeaks
DBPeaks is a Python package for identifying peaks in CRAC/CLIP data.
It uses pyCRAC tools together with bedtools and DESeq2 to identify reproducible protein-RNA interaction sites (peaks) in CLIP/CRAC data. It was specifically designed to compare CLIP/CRAC datasets generated under different conditions to identify differential binding sites.
DBPeaks was used in our recent Esteban-Serna nucleic acids research paper (Esteban-Serna et al. 2025) to identify differential binding sites (see Figure 1F).
Code repository: https://git.ecdf.ed.ac.uk/sgrannem/dbpeaks
Reference:
Sofia Esteban-Serna, Tove Widen, Mags Gwynne, Iseabail Farquhar, Michael Duchen, Peter S. Swain and Sander Granneman#.
A transcription termination mechanism for maintaining homogeneous protein expression.
Nucleic Acids Research. 2025 Nov 13;53(21):gkaf1199. DOI: 10.1093/nar/gkaf1199
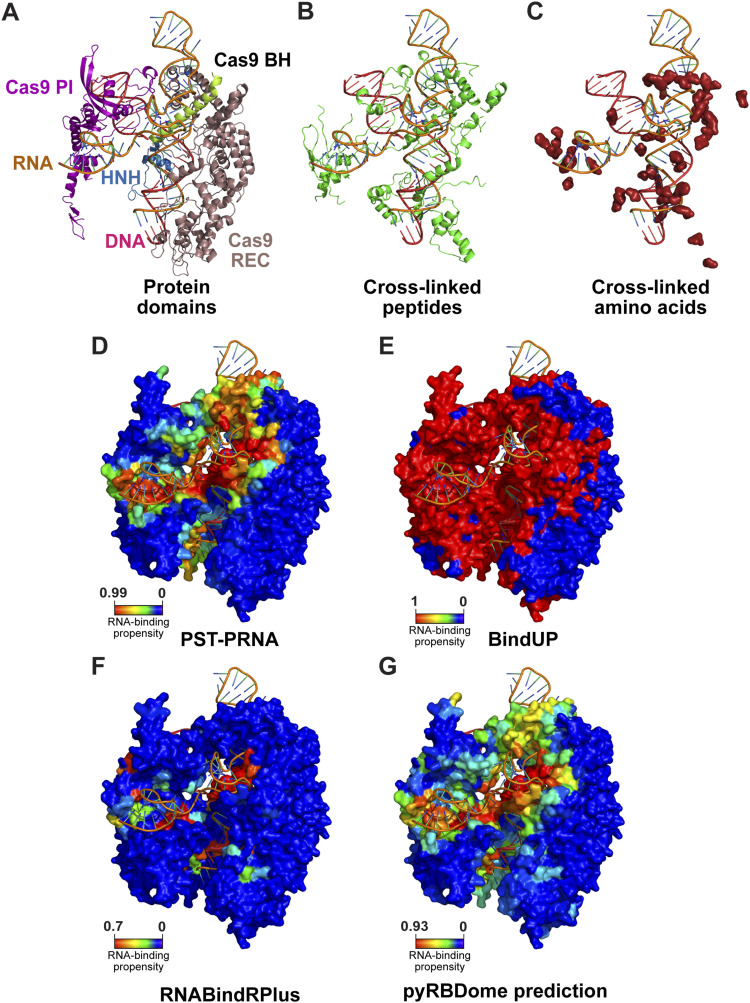
pyRBDome
pyRBDome is a computational pipeline developed to analyze RNA-binding proteome data. Over the past few years, we and others have produced a large number of proteomics datasets that revealed many putative RNA-binding proteins (RBPs). However, it was unclear what fraction of these putative RBPs were genuine RNA-binders.
To address this, we developed pyRBDome to enhance the experimental data. It is a comprehensive pipeline that enables users to identify putative RNA-binding regions within proteins and perform statistical analysis to determine if peptides identified in these proteomics studies are indeed bona-fide RNA-binding regions.
pyRBDome was also used in the following 2025 publications:
- RNMT: L. A. Hepburn, Sander Granneman, V. H. Cowling and J. Clara Silva. RNMT is recruited to RNA by interaction with RNA G-quadraplexes. bioRxiv. 2025 Nov 16. DOI: 10.1101/2025.11.16.688685
- NF45-NF90: Sophie Winterbourne, Uma Jayachandran, Juan Zou, Juri Rappsilber, Sander Granneman, Atlanta G. Cook. Integrative structural analysis of NF45-NF90 heterodimers reveals architectural rearrangements and oligomerization on binding dsRNA. Nucleic Acids Research. 2025 Mar 20;53(6):gkaf204. DOI: 10.1093/nar/gkaf204
Code repository: https://git.ecdf.ed.ac.uk/sgrannem/pyRBDome_Core
Reference:
Liang-Cui Chu*, Niki Christopoulou*, Hugh McCaughan*, Sophie Winterbourne, Davide Cazzola, Shichao Wang, Ulad Litvin, Salomé Brunon, Patrick J.B. Harker, Iain McNae and Sander Granneman. pyRBDome: A comprehensive computational platform for enhancing RNA-binding proteome data. Life Science Alliance. 2024; 7 (10) e202402787; DOI: 10.26508/lsa.202402787
Installation
We strongly recommend installing pyRBDome in a separate virtual environment to avoid conflicts with other packages.
Create a virtual environment and install from GitLab:
# Create and activate a virtual environment
python3 -m venv pyrbdome_env
source pyrbdome_env/bin/activate
# Clone and install
git clone https://git.ecdf.ed.ac.uk/sgrannem/pyrbdome.git
cd pyrbdome
pip install .diffBUM-HMM
diffBUM-HMM is a robust statistical modeling approach for detecting RNA flexibility changes in high-throughput structure probing data.
It provides a statistical framework for analyzing data from high-throughput RNA structure probing experiments (such as SHAPE-MaP, DMS-MaPseq, etc.) to identify significant changes in RNA structure flexibility between different conditions.
Code repository: https://github.com/sgrann/diffBUM-HMM
Reference:
Paolo Marangio*, Ka Ying Toby Law*, Guido Sanguinetti# and Sander Granneman#.
diffBUM-HMM: a robust statistical modeling approach for detecting RNA flexibility changes in high-throughput structure probing data.
Genome Biology, 2021. PMCID: PMC8157727 DOI: 10.1186/s13059-021-02379-y
# corresponding authors, * These authors contributed equally